Pixel-by-pixel control of light exposure brings benefits to live-cell image.
Dr. Jennifer L. Peters, Nikon Instruments Inc.
Fluorescence imaging of live cells and tissues is a powerful technique that is increasingly finding new applications in the biological sciences.1 However, photobleaching and phototoxicity can present significant challenges for many types of live-cell imaging experiments, including many laser-based applications. Photobleaching — the fading of fluorophore molecules — can seriously degrade image quality and limit the amount of time useful data can be collected from live cells.2 Phototoxicity also is a serious concern: Excitation light can cause generation of free radicals that react with easily oxidizable compounds, including nucleic acids and proteins, and can ultimately lead to cell death.3
In live-cell imaging, various strategies are used to maximize fluorescence signal while minimizing incident illumination and the resulting photodamage. For example, it has become routine to use highly sensitive detectors and efficient optics to collect as many of the resulting fluorescent photons as possible, thus reducing the amount of illumination needed to generate an image with sufficient signal to noise.
In controlled light exposure microscopy (CLEM), however, a unique new approach is used to reduce the excitation light incident on the sample. The technique was developed by Erik Manders and his team at the University of Amsterdam in the Netherlands.4
In conventional laser scanning confocal microscopy, every pixel is illuminated equally, even in region of interest scans. As a result, the sample is illuminated unnecessarily at pixels where there are no fluorophore molecules present, potentially causing photobleaching and phototoxicity above and below the plane of focus. Furthermore, pixels with high fluorophore concentration can be illuminated longer than is necessary, producing an image with higher than necessary signal to noise or oversaturating the image at that pixel. This results in unnecessary and potentially damaging laser exposure, as well as a limited or clipped dynamic range.
The basic principle of CLEM is the use of “intelligent scanning” to reduce laser excitation pixel by pixel. Laser light is applied with varying intensity across the image, illuminating only where needed and for only as long as necessary.
It works by employing a two-part strategy: First, when no fluorescence signal is detected at a particular pixel, the illumination of that pixel is stopped. Second, when the fluorescence emission at a particular pixel is saturated, the illumination at that pixel is similarly ended.

Figure 1. The images of fluorescence microspheres alone (left) and with an overlaid saturation indicator (right) illustrate the strategy used by controlled light exposure microscopy (CLEM) to reduce laser excitation pixel by pixel.
In Figure 1, fluorescent microspheres illustrate this point. The left image shows the microspheres, and in the right image, blue indicates pixels with no detectable fluorophore, whereas red indicates saturated pixels. In conventional confocal imaging, all of these pixels are illuminated equally, but with the intelligent scanning of CLEM, pixels in the red and blue regions are illuminated less, reducing photobleaching and phototoxicity while having no effect on imaging quality.
This technique has been implemented in Nikon’s C1-CLEM system. The instrumentation needed for CLEM imaging is relatively simple (Figure 2). Compared with a traditional confocal system, it requires the addition of an electronic unit that provides feedback from the confocal detector’s analog-to-digital electronics to the acousto-optical modulator (AOM).
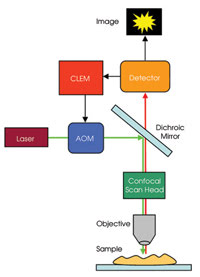
Figure 2. The CLEM system requires the addition of an electronic unit to a confocal laser scanning microscope. The unit provides feedback from the confocal detector’s analog-to-digital electronics to the acousto-optical modulator (AOM).
The unit’s high-speed electronics work by monitoring the first portion of the pixel period (default is 20 percent, or the value can be selected). The AOM reduces the laser power to zero with submicrosecond temporal resolution if the fluorescence signal is above or below selectable threshold values. If the signal is below the threshold value, there is no signal at that pixel, whereas if the signal is above the high threshold value, saturation will be reached at that pixel. By this mechanism, the sample is illuminated only for as long as necessary at locations where it is needed, extending the relative dynamic range of the image and reducing the effects of photobleaching.
Manders and colleagues4 have used the system for time-lapse imaging of GFP-expressing HeLa cells. Cells were imaged every 35 seconds at two stage positions: one with CLEM and the other without it. Figure 3 shows the fluorescence overlaid onto the phase contrast images for several time points during the experiment. In the non-CLEM series of images, after 26 minutes the cells show blebbing of the membrane, which is an early sign of cell death. By the end of the experiment, nearly all of the cells have rounded up and are apoptotic.
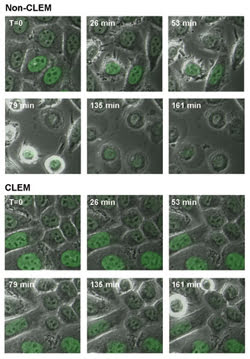
Figure 3. GFP-labeled HeLa cells, imaged every 35 s for more than 4 h with CLEM (bottom) and without CLEM (top), demonstrate the decrease in phototoxicity that can be achieved with the system.
In contrast, when CLEM is used, no signs of cell death are seen in any of the cells until near the end of the experiment. Manders and his coworkers found that they could image cells approximately six times longer with the system than with conventional laser scanning confocal imaging. This experiment suggests that the reduction of laser illumination provided by the system’s intelligent scanning greatly reduces the generation of free radicals, delaying apoptosis and allowing cells to be imaged longer.
In addition to reducing phototoxicity, it also can be demonstrated that the system reduces photobleaching. In a second experiment, HeLa cells were imaged with and without CLEM (Figure 4). In each case, 35 stacks of 21 images each were taken, and a single image plane from the first and last stack in each sequence of stacks is shown.
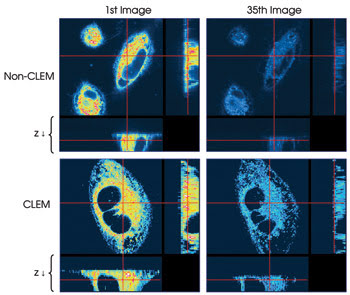
Figure 4. A time-lapse experiment where 35 image stacks of 21 images each were taken with (bottom) and without CLEM (top) demonstrates the possible reduction in photobleaching. Single image planes from the beginning and end of each series of image stacks are shown.
Whereas the image taken from the initial stack in each sequence shows nearly the same intensity, in the image from the last non-CLEM stack, essentially no signal remains. In contrast, using the CLEM system results in a much brighter image at the end of image collection of the 35 stacks. Whereas in the non-CLEM experiment the image intensity has been reduced by one-half after the collection of 10 stacks, with CLEM imaging a similar reduction of one-half is not seen until stack 25, suggesting that it takes 2.5 times longer for photobleaching to occur.
Additionally, the technique expands the dynamic range of the measurement. Typically in laser scanning confocal microscopy, the power of the laser excitation is adjusted in regions where the fluorophore concentration is low so that there is enough intensity emitted from the fluorophore to discern structures with sufficient signal to noise. Consequently, because regions with high fluorophore concentration are illuminated equally, those regions can become saturated, limiting the dynamic range.
Because the system illuminates the sample nonuniformly, by attenuating the laser illumination at pixels where the signal is higher than needed to saturate, the dynamic range is increased, and the problem of oversaturation is eliminated. This is especially significant when collecting Z-series or -stacks of images.
In a Z-series, planes near the surface often are oversaturated because of the need to keep laser power high enough to overcome losses to scatter deeper into the specimen. With the intelligent scanning, planes deeper in the specimen receive more laser illumination without the planes near the surface becoming oversaturated, expanding the dynamic range and improving image quality. The ability to collect an unsaturated Z-stack of images in a thick specimen preparation with increased dynamic range is an important improvement to current confocal image acquisition methods.
With the explosion in the availability of fluorescent proteins and other probes for a wide variety of applications, live-cell imaging has become an indispensable tool in cell biology research.5 The system addresses many of the challenges of live-cell imaging. Manders and others have demonstrated that photobleaching and phototoxicity are both decreased several-fold with CLEM, allowing specimens to be imaged longer than ever before possible.4 This in turn allows meaningful data to be collected for much longer, and it makes possible experiments that were previously extremely technically challenging, such as long-term studies of protein expression, cell growth, motility or apoptosis, to name a few.
Furthermore, the reduction of photobleaching and the extension of dynamic range achieved with the system are beneficial when imaging fixed cells and tissues, for example with immunolabeled samples, and especially in the case of image stacks. For microscopists performing a wide variety of experiments, the technique is proving to be an exciting new tool for confocal imaging, with applications in many areas of biological research.
Meet the author
Jennifer L. Peters is an advanced biosystems applications scientist at Nikon Instruments Inc. in Melville, N.Y.; e-mail: [email protected].
References
1. D.J. Stephens and V.J. Allan (2003). Light microscopy techniques for live cell imaging. SCIENCE, Vol. 300, pp. 82-86.
2. T. Bernas et al (2004). Minimizing photobleaching during confocal microscopy of fluorescent probes bound to chromatin: role of anoxia and photon flux. J MICROSC, Vol. 215, pp. 281-296.
3. R. Dixit and R. Cyr (2003). Cell damage and reactive oxygen species production induced by fluorescence microscopy: effect on mitosis and guidelines for non-invasive fluorescence microscopy. PLANT J, Vol. 36, pp. 280-290.
4. R.A. Hoebe et al (2007). Controlled light-exposure microscopy reduces photobleaching and phototoxicity in fluorescence live-cell imaging. NAT BIOTECHNOL, Vol. 25, pp. 249-253.
5. J. Lippincott-Schwartz and G.H. Patterson (2003). Development and use of fluorescent protein markers in living cells. SCIENCE, Vo. 300, pp. 87-91.