Understanding attosecond-scale phenomena may enable electronics 100,000 times faster than today’s devices.
Reinhard Kienberger, Max Planck Institut für Quantenoptik
New insight into ever-smaller structures of matter and their ever-faster dynamics holds great promise for pushing the frontiers of many fields in science and technology. The speed of electronic processes is determined by the separation of the energetic states of electrons. In atoms, the binding energy of electrons is on the order of tens to hundreds of electron volts, which explains why electronic processes occur on an attosecond timescale. One attosecond (10–18 s) is the billionth part of a nanosecond, which is itself the billionth part of a second. Put more picturesquely, one attosecond compares to one second approximately as one second to the age of the universe.
In pump/probe experiments, one pulse (of light, of x-rays, of electrons, etc.) triggers a process, and another pulse with an adjustable delay probes the subsequent temporal evolution of the process. This has turned out to be the most direct approach to time-domain investigations of fast-evolving microscopic processes.
Accessing atomic and molecular inner-shell relaxation processes, valence-band electron wave packet dynamics, electron-electron interactions or atomic-scale charge transport in real time requires subfemtosecond temporal resolution. The emergence of intense waveform-controlled few-cycle near-infrared laser pulses and isolated subfemtosecond extreme-ultraviolet (XUV) pulses available for pump/probe experiments has opened the door for direct time-domain access to electron motion on the atomic (i.e., subnanometer) scale. These tools, along with the technique of laser-field-controlled XUV photoemission (to be described below), have permitted real-time observation of intra-atomic electronic motion in several proof-of-concept experiments performed on isolated atoms in the gas phase and mark the birth of attosecond science and time-resolved atomic transient recording.
Recently, we applied the tools of attosecond physics in an experiment on a solid’s surface, extending attosecond physics to solid-state and surface science. Fast electron dynamics on solid surfaces are of broad scientific interest and are pertinent to the development of many technologies, including, but definitely not limited to, semiconductor electronics, optoelectronics and photovoltaics. However, exploration of these dynamics remains a significant challenge for experimentalists in the laboratory today. Whereas substantial progress has been made in observing coherent and incoherent atomic motion through time-resolved x-ray diffraction and through the development of fourth-generation femtosecond x-ray sources, complementary studies of attosecond electronic motion in solids and on surfaces have — until now — remained out of reach.
Atomic transient recording
Why is it so important to extend atomic transient recording to solids, and what are the first results found with this technology?
The controlled transport of electric charge by electrons through nanoscale electric circuits forms the basis of modern electronics. Motivation for developing faster electronics comes from many directions. Faster computers and more sensitive instruments will allow more reliable prediction of natural disasters and deeper insight into the workings of nature by ever-more-sophisticated modeling. Ultrahigh-speed communications systems may, one day, permit specialists to perform remote surgery and will make health care more efficient in many other ways. Further possible implications are limited only by one’s imagination.
In state-of-the-art electronic circuits, electrons are driven by microwave voltages, which are capable of switching current on and off in a fraction of a nanosecond. The switching time determines how fast the computer performs calculations. Ultimately, the rapidity of switching is limited by the time it takes for the electrons to travel through the structures that guide and control them. Smaller structures lead to faster switching speeds and a higher density of information flow.
The quest for ever-smaller nanostructures in solid-state electronics and for atomic assemblies in molecular electronics is driven by these simple considerations. The distance between neighboring atoms in a crystal lattice or in a molecule constitutes the smallest possible distance for channeling and switching current. It takes only attoseconds for electrons to travel these atomic-scale distances, implying the feasibility of switching current more than a trillion times a second in atomic-scale circuitry. If achieved, this would result in the emergence of petahertz electronics, in which the direction of electric current can be changed several trillion times per second. This is about 100,000 times higher than permitted by today’s electronics.
The ultimate speed in electronics would be limited only by the time required for an electron to travel between neighboring atoms. An essential first step on the long journey toward that speed is the development of techniques to capture electronic charge transport in atomic-scale structures on the attosecond timescale. We recently completed a proof-of-principle experiment in which we observed attosecond electric charge transport across several atomic layers near the surface of a crystalline solid in real time.1 Figure 1 shows the setup of an experiment where the different speeds of electrons in a tungsten crystal are investigated in an XUV-pump/IR-probe experiment.
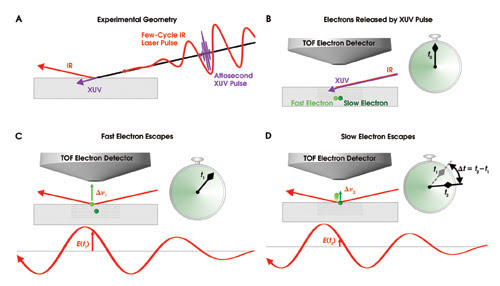
Figure 1. In an attosecond extreme-ultraviolet (XUV) pump/IR probe experiment, the velocities of electrons in a solid are determined. In panel (A), a few-cycle waveform-controlled IR laser pulse and an attosecond XUV pulse are incident on a tungsten crystal. The XUV pulse penetrates the crystal. In panel (B), the attosecond XUV pulse is absorbed into the crystal, setting free two types of electrons: fast ones responsible for conduction and slow ones originating from the vicinity of the atomic cores. The electrons freed in this fashion within several layers below the crystal surface likely reach the surface without energy-reducing collisions and, thus, escape the crystal. Panels (C) and (D) capture the instances when the fast and slow electrons reach the surface and subsequently are sped up and slowed down, respectively, by the IR laser pulse. The laser field-induced changes in their velocities differ significantly because of the disparity in strength of the laser electric fields E(t1) and E(t2) at t1 and t2. The changes in the electrons’ velocity, denoted by Δv1 and Δv2, can be measured by a time-of-flight (TOF) electron detector, from which the time elapsing between the arrival of the two types of electrons on the surface, vt = t2 – t1, can be determined with attosecond accuracy.
Working with colleagues from Bielefeld University in Germany, we directed a 300-attosecond XUV pulse, along with an IR laser pulse comprising a few well-controlled oscillation cycles of its electric field, onto the surface of a tungsten crystal. The attosecond pulse penetrated the tungsten crystal, where the XUV photons were absorbed, liberating photoelectrons. There were two types of photoelectrons: loosely bound ones responsible for conduction in the metal and ones that had been tightly bound to the atoms forming the crystal lattice. Once the electron was liberated, its initial velocity depended on the difference between the XUV photon’s energy and the electron’s binding energy; that is, the original law of photoemission as described by Einstein more than a century ago still applies, even on an attosecond timescale.
Once at the surface, the electrons feel the electric field of the synchronized IR pulse changing their speed, depending on when they escape the solid. From the measured changes in the slow and fast electrons’ initial velocity, attosecond-scale differences in the arrival time of the electrons can be determined unambiguously. By changing the electrons’ velocity in a controlled fashion, the rapidly oscillating laser field serves as a stopwatch with attosecond resolution.
Let us now have a look at how attosecond XUV pulses are generated from IR laser pulses — and therefore are perfectly synchronized — and how the two pulses (XUV and IR) can be used in pump/probe experiments.
High-order harmonic generation
One very important tool of attosecond technology is a source of radiation having two characteristics: (i) temporal and spatial coherence and (ii) high energy — in other words, coherent, ultrafast light sources in the soft and hard x-ray regime.
One promising way to generate coherent radiation at short wavelengths is to convert laser light to higher harmonics. Excellent coherence and relatively low pump-energy requirements make high-harmonic generation attractive for developing a laboratory short-wavelength source that can provide a collimated, laserlike beam.
The harmonic conversion is performed in a nonlinear medium, typically a gas where the electric field of the laser pulses interacts with electrons. In a semiclassical view, the atomic potential binding the electrons to the core is “bent” (see Figure 2a) by the laser’s electric field — especially at the instants of its extreme values — such that an electron can tunnel through and escape from the atom.
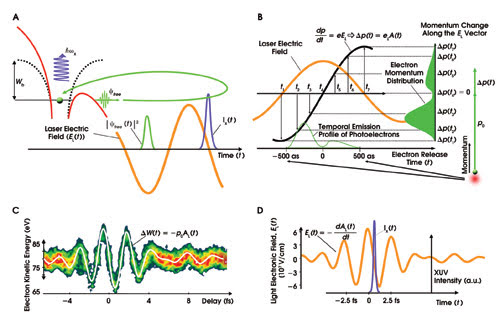
Figure 2. (A) The upper-left portion of this figure shows an electron (green sphere) in its normal atomic potential well (broken black lines). Wb is the binding energy, or work function. The walls of the well are “bent” (red lines) by the laser’s electric field (orange sinusoidal trace), and the electron tunnels to freedom (green arrow). Initially — when the laser field is shown as negative here — the laser field accelerates the electron away from the atom. However, when the field reverses, it slams the electron back into the atom, releasing energy in the form of a high-energy photon (purple wave function). The green loop represents the spatial journey of the electron, driven by the laser field. (B) Similar to the way a streak camera maps a temporal profile to a spatial profile, the attosecond-streaking process maps a temporal profile to a momentum profile. (C) The spectrogram, a series of streaked electron spectra recorded at various instants of time, can be used to fully reconstruct the near-IR laser field. (D) Also, one can reconstruct all parameters of the process setting free the measured electrons: for photoelectrons, this is the XUV pulse duration; for secondary electrons, this is the process setting them free (e.g., Auger process).3

The electron initially is driven away from its parent atom (ion) by the laser electric field. Because the latter is oscillating, it is later driven back to the atom. The collision creates a photon with the entire energy that the electron has accumulated during its journey. In this manner, XUV or soft-x-ray photon energies can be achieved. The laser electric field interacts with millions of atoms in a perfectly synchronized way so that the XUV emission takes place every half-cycle of the field, generating a significant number of photons each time. In other words, high-harmonic generation creates a train of subfemtosecond or even attosecond bursts of coherent short-wavelength radiation.
The pump/probe technique
For a singular triggering of a process and unambiguous probing, scientists need single, isolated pulses. The success of the generation of isolated attosecond pulses requires producing intense femtosecond few-cycle IR laser pulses with controlled waveforms as a driver for harmonic generation (Figure 2a). The driving IR laser field must be extremely short, lasting for only a few optical cycles, of which only one cycle should have dominant amplitude. State of the art for the duration of pulses in the submillijoule regime — which is needed for high-harmonic generation — is about 1.5 cycles of the carrier wave. For visible light — for example, at the central wavelength of Ti:sapphire lasers around 750 nm — one oscillation cycle lasts about 2.5 fs. This means that laser pulses having a duration of about 3.5 fs are available.2
The highest-order harmonics are produced near the intensity peak of these laser pulses because the recombination energy of the electrons is highest when the laser field is largest. If the pulses reach a duration of a few light-field oscillation cycles, the highest-order harmonics (cutoff) merge to a continuum. By using a few-cycle driver and passing the harmonics through a filter that transmits only the highest frequencies, investigators can filter out everything except radiation produced near the peak of the driving pulse.
Multilayer mirrors with a reflectivity at the photon energy corresponding to the cutoff of the high-harmonic radiation can provide the necessary filtering. For historical reasons — these mirrors were first used in lithography — mirrors reflecting ~13 nm (~100-eV photon energy) are well developed. They support a bandwidth of up to 30 eV, at a reflectivity of a few percent. Efforts in source development to reach higher photon energies by high-harmonic generation, as well as by tailoring multilayer mirrors for higher energies, are being undertaken by several research groups around the world.
The method of filtering cutoff harmonics can generate single isolated XUV/x-ray pulses of 100-attosecond duration, and the latest experimental results have reached this limit. Because these processes are very sensitive to the waveform of the laser pulse, it is necessary to stabilize and control the carrier-envelope phase. Only “cosine” pulses — pulses whose most prominent field peak coincides with the envelope maximum — have the potential to produce single bursts. The recent demonstration of phase-stabilized intense few-cycle laser pulses2 allows the generation of cosine pulses for the reproducible production of single attosecond pulses on a daily basis.
Our investigation of surface science relies on the pump/probe technique, using XUV pulses created by high-harmonic generation as the pump and the driving laser pulse as the probe, or vice versa. We call our system an atomic transient recorder3; it constitutes the main tool of attosecond physics (Figure 2b). The basic prerequisite, namely the generation and measurement of isolated subfemtosecond XUV pulses synchronized to a strong few-cycle IR pulse with attosecond precision, opens a route to time-resolved inner-shell atomic as well as to molecular spectroscopy with present-day sources.4
Streaked spectrum
Electrons released from a sample by an XUV pulse parallel to the direction of the IR laser electric field (the orange trace in Figure 2b) suffer a change of their initial momenta that is proportional to the vector potential of the laser field (black line) at the instant of release, mapping the temporal intensity profile of the emitted electrons — and, hence, of the ionizing subfemtosecond XUV pulse — into a corresponding final momentum and energy distribution of electrons. Because the energy distribution of the photoelectrons changes its shape — because of the laser field — it is called “streaked spectrum.”
An atomic-transient spectrogram is compiled by measuring a series of streaked photoelectron (or secondary — e.g., Auger — electron) spectra with a time-of-flight electron detector (as shown in Figure 1), recorded as a function of time delay between the XUV and IR streaking field.
Figure 2c shows such a spectrogram of streaked spectra of photoelectrons released from neon atoms by a single subfemtosecond XUV pulse (hωx ≈ 95 eV) recorded for a series of delays between the XUV pulse and near-IR field. The laser field causes only a moderate broadening (streaking) of the electron spectra, with its main effect being the shift of the center of the electron spectrum. In the limit of large initial kinetic energy of the electrons, this shift is linearly proportional to the vector potential of the field at the instant of impact of the XUV pulse (the white line in Figure 2c).
Important characteristics of the emitted electron wave packets, including their duration and frequency sweep, or “chirp,” can be determined from the spectra. Figure 2d shows the electric field of the IR laser field and the intensity of the XUV pulse as retrieved from the streaking spectrogram shown in Figure 2c. The measurements confirm that the few-cycle waveform must be nearly cosine-shaped to produce a single subfemtosecond pulse and reveals a near-Fourier-limited XUV pulse of a duration of 250 attoseconds. The resolution of our atomic transient recorder depends on the duration of the XUV excitation, the gradient of the streaking IR field and the signal-to-noise ratio in the photoelectron spectra.
In a solid such as tungsten, one gets two parallel spectrograms originating from two types of electrons — conduction and core electrons — being emitted at different energies. Both spectrograms show the change in electron energy corresponding with the evolution of the electric field of the infrared pulse. By careful examination of the measured data in Figure 3, we determined that the loosely bound conduction electrons were emitted approximately 100 attoseconds earlier than their tightly bound counterparts. This delay indicates that, inside the tungsten crystal, the freed conduction electrons travel twice as fast as those liberated from states localized near the atomic cores.
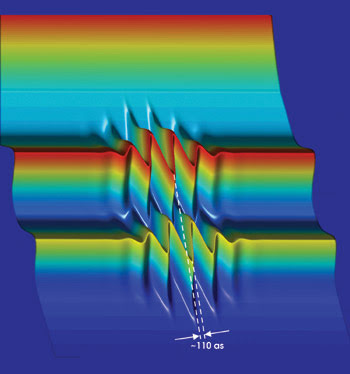
Figure 3. The two spectrograms of conduction-band electrons and core electrons in tungsten were captured simultaneously and are parallel on the (vertical) energy axis because of the different emission energies of the two types of electrons. Both show the electrons’ change in energy caused by the changing electric field of the IR pulse shining on the surface when the electrons escape. The tiny but resolvable shift of the spectrograms on the (horizontal) time axis indicates the delayed arrival of the respective electrons at the solid surface.
These experiments on a solid surface demonstrate the technical ability of measuring electronic charge transport across atomic layers in real time with attosecond temporal resolution. Future measurements of this type will allow for research into structures and technologies for speeding up state-of-the-art electronics by several orders of magnitude. Real-time control of attosecond-scale electronic processes in condensed-matter systems will enable exploration of the ultimate limits of the aforementioned technologies. It will identify ways of approaching these limits; for example, via molecular electronics, plasmonics or other technologies.
Acknowledgment
The author acknowledges funding from the Sofja Kovalevskaja Award granted by the Alexander von Humboldt Foundation.
References
1. A.L. Cavalieri et al (2007). Attosecond spectroscopy in condensed matter. NATURE, Vol. 449, pp. 1029-1032.
2. A.L. Cavalieri et al (July 2007). Intense 1.5-cycle near infrared laser waveforms and their use for the generation of ultra-broadband soft-x-ray harmonic continua. NEW JOURNAL OF PHYSICS, Vol. 9, p. 242.
3. R. Kienberger et al (2004). Atomic transient recorder. NATURE, Vol. 427, pp. 817-821.
4. E. Goulielmakis et al (2007). Attosecond control and measurement: Lightwave electronics. SCIENCE, Vol. 317, pp. 769-775.
Meet the author
Reinhard Kienberger is the leader of the independent junior research group Attosecond Dynamics at Max Planck Institute of Quantum Optics in Garching, Germany.