Researchers at the University of California, San Francisco (UCSF) are combining multiple laser excitation wavelengths in total internal reflection fluorescence (TIRF) microscopy to investigate the binding dynamics of individual actin filaments.
Dan Callen, Coherent Inc.
TIRF microscopy provides a unique method of imaging isolated molecules and complexes in vitro. Additionally, the use of sensitive, low-noise cameras enables researchers to study this behavior in real time. A new plug-and-play method of combining several fiber-delivered, digitally modulated lasers into a single instrument, such as a TIRF microscope, now enables multiple labeled proteins to be imaged pseudosimultaneously at high frame rates. This article explores how multiwavelength excitation is being combined with TIRF microscopy in the laboratory of Dr. Dyche Mullins, a professor at UCSF, and how it’s being used to gain new insights into complex biochemical interactions that control the stability and function of actin filaments.
TIRF microscopy in single-filament studies
The Mullins Lab, located at UCSF’s Mission Bay campus, is widely recognized as a leading authority on the study of actin filaments. The protein filaments are fundamental to many processes in virtually every eukaryotic cell — they act as structural elements that enable movement of internal cargoes, amoeboid cell migration, cell division, etc. With these filaments playing so many different roles, it is not surprising that their combination of growth, branching, aggregation and movement involves many subtle control options, which are mediated by a range of different proteins. Sam Lord, the Mullins Lab’s microscope specialist, said, “One area of our research is studying how various proteins bind to actin filaments to enable aggregation, branching and other actions, and more specifically, how yet another set of proteins modulates these binding processes. Obviously, we do bulk studies in a cuvette that reveal overall kinetic data about these binding processes but we also want to image these processes in real time to study the structural biochemistry.” In order to do so, the lab uses TIRF microscopy to observe single actin filaments.
This process involves excitation light that is introduced into the sample region through either a glass slide or a cover slip. The microscope’s optics are configured so that the light hits the glass/sample interface beyond the critical angle, meaning that all of the light will undergo total internal reflection (TIR). However, even with TIR, some of the light’s electric field, called the evanescent wave, penetrates into the sample by an incredibly short distance — typically around 100 nm — beyond the interface. This means that TIRF microscopy can be used to selectively excite fluorescence in molecules and complexes that are adhered to the interface. However, because the light does not penetrate into the bulk (i.e., background) sample region, this methodology will not excite fluorescence from the huge backdrop of molecules freely floating within this medium.
TIRF microscopy is thus a 3D-resolved imaging technique. Its X-Y resolution is limited only by diffraction and/or the camera resolution, but the Z-axis sampling depth is much smaller than the diffraction limit. If there is sufficient signal for fast frame acquisition speeds, the important fourth dimension — time — enables dynamic processes, such as actin filament-protein binding, to be observed on a single filament or on a network of filaments, in real time.
In principle, both laser and nonlaser light sources may be used for fluorescence excitation in such TIRF-based applications. However, for experiments with naturally low signal levels, such as single-molecule monitoring, a laser beam’s extreme brightness is a critical advantage. In particular, a laser’s unique spatial brightness means that it is relatively simple to collimate and subsequently focus the beam into the sample with a narrow range of incidence angles, avoiding excitation of the bulk sample.
Through-objective TIRF microscopy
All TIRF microscope setups are based on one of two basic approaches: through-objective lens geometry or the prism-based method. In the former approach, light is directed in an off-axis geometry through an oil-immersion microscope objective so that the angle of incidence at the coverslip/sample interface is greater than the critical angle, as is shown schematically in Figure 1.
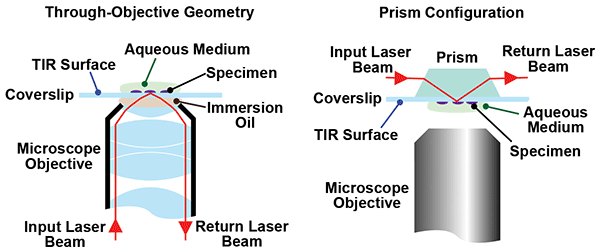
Figure 1. In TIRF microscopy, excitation light beyond the critical light is completely reflected. The evanescence of the light field at the refractive interface penetrates into the sample by about 100 nm, causing selective excitation of molecules and complexes adhered to this interface. TIRF microscopes are available with a choice of either through-objective excitation or prism excitation options.
In the prism-based method, the orientation of the sample is reversed with respect to the imaging objective. A light beam is introduced to the sample through a prism attached to the cover slip; the geometry of the prism ensures that the incidence angle at the sample is greater than the critical angle.
Depending on the type of experiment being performed, there are both advantages and disadvantages to each of the above methods. For example, the prism method limits physical access to the sample. As Lord explained, the Mullins Lab uses a
Nikon microscope in the through-objective configuration with a very high numerical aperture (NA = 1.49) for several reasons. “For single-molecule studies, fluorescence signal strength is always a major challenge, particularly since we are following processes that need fast frame rates. So, we need a high-NA objective with a small working distance to maximize light collection efficiency. These objectives require a coverglass of precise thickness and the sample near the top of the coverslip to minimize aberrations.” Lord also stated that caution must be taken so the team does not introduce scattering and other losses due to viewing fluorescence through the bulk of the sample.
Multiple, simultaneous laser wavelengths
As has been noted, the mechanisms controlling the binding of regulatory proteins to actin filaments are quite complex. To better understand these processes, the Mullins Lab increasingly has been using sophisticated, multiwavelength TIRF-based experiments. In order to image multiple fluorophores, Lord explained, “We can use either multiple sequenced lasers or a scope equipped with multiple cameras — we have setups for both arrangements.” He continued, “Multiple excitation wavelengths that sequence at high rates enable us to selectively image multiple, differently labeled targets using a microscope equipped with a single high-sensitivity camera, and ensures near-perfect image registration.”
When using multiple lasers, the two technical challenges are to perfectly coalign the lasers into the microscope objective and then to be able to switch between different wavelengths. In order to follow fast binding processes in real time, researchers typically must switch wavelengths between alternate camera frames to build up pseudosimultaneous (i.e., interleaved) videos at two or sometimes three laser wavelengths. This switching must be performed with no undesirable dead time (i.e., shifts in the beam path) and without using mechanical shutters or a complex and costly approach, such as an acousto-optic tunable filter.
Lord notes, “As recently as five years ago, we simply didn’t have low-cost options to conduct single molecule studies using multiple laser wavelengths pseudosimultaneously at the requisite frame rates (30 fps) in order to follow critical binding processes.” He added, “Digitally controllable diode or solid-state lasers, hardware sequencing electronics and quad-band optical filters make it possible to achieve nearly simultaneous multicolor imaging with a single camera.”
In 2014, the lab acquired several digitally modulatable smart lasers to enable multiwavelength TIRF microscopy. These lasers included Coherent’s fiber-pigtailed OBIS FP modules that operate at 488, 561 and 640 nm. The lab also acquired the OBIS Galaxy, which enables simple plug-and-play combining of up to eight fiber-coupled lasers into one, single-mode output fiber. As was detailed by Coherent’s Dan Callen and Matthias Schulze in BioPhotonics’ November 2014 issue (“Laser Combiner Enables Scanning Fluorescence Endoscopy,” www.photonics.com/A56915), this passive module enables lasers to be added or subtracted (i.e., hot-swapped) to any fiber-coupled instrument or setup in a few minutes or less via standard fiber connectors, such as FC/UFC and FC/APC connectors.

Figure 2. The OBIS Galaxy (shown with the top cover removed) allows plug-and-play combining of up to eight separate fiber coupled lasers into a single output fiber. Courtesy of Sam Lord.
The timing hardware setup at the Mullins Lab is very simple in design due to the fact that these smart lasers support direct digital modulation. In each experiment, the frame rate is set by the microscope’s high-sensitivity camera, which is an Andor DU897. The camera’s TTL output trigger pulses are processed in either a programmable Arduino board or an ESio controller, which then directs TTL pulses to fire one of the three lasers without any hardware or software delays. Alternating wavelengths typically are used in most experiments, although any sequence of wavelength frames easily can be programed using the Arduino and Micro-Manager software.1
According to Lord, the flexibility of this arrangement supports future experimental setups that have even greater levels of complexity. In particular, he added, “We may well add a 405-nm laser option in the near future. If/when this arrives, we can simply plug it in and we are ready to go.”
Investigating modulation of actin binding processes
In the team’s work on the binding of actin filaments, this flexible TIRF setup enables the Mullins Lab to conduct experiments with several different approaches. For example, in typical two-wavelength experiments, the actin filament is labeled with one fluorophore, and the protein of interest is labeled with another fluorophore. The protein fluorophore only appears in the TIRF-produced images if/when it binds to the actin sitting on the cover slip. One use of the third wavelength is to image a second protein, which is labeled with a different fluorophore. The image sequences then may reveal, for example, whether the proteins are interspersed at different sites on the filament, or whether the second protein promotes filament growth or branching from a new site. Or, it may reveal that the second protein competitively displaces the first.
In a recently published study,2 Mullins Lab researchers used their multilaser TIRF setup to investigate the details of control mechanisms associated with the binding of tropomyosins to actin filaments. Tropomyosins are coiled-coil proteins whose known functions are to bind actin filaments and thereby regulate multiple cytoskeletal functions — including actin network dynamics near the leading edge of motile cells.
Mullins explained, “The binding of tropomyosins to actin filaments is known to be fundamentally important in actin dynamics. But, we do not yet fully understand how this binding is regulated, especially near the leading edge of migrating cells. Why, for example, are filaments in the lamellum coated with tropomyosin while filaments in the adjacent lamellipod are not?” (Lamellum and lamellipod are distinct, actin-based substructures involved in cell migration.) He went on to state that, prior to his team’s latest studies, previous research demonstrated that tropomyosins inhibit actin nucleation by the Arp2/3 protein complex and that this, in turn, prevented filament severing by the protein cofilin.3,4 “So, we have recently used TIRF and other methods to investigate if and how the Arp2/3 complex and cofilin in turn modulate the binding of tropomyosins to actin filaments,” he said.
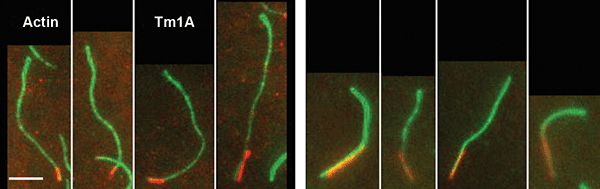
Figure 3. TIRF images showing Tm1A binding preferentially to the pointed end of single actin filaments. The red signal is from Cy5 labeled Tm1A fluorescence excited at 640 nm, and the green signal is due to Alexa 488 labeled actin excited at 488 nm. Courtesy of J.Y. Hsiao, L.M. Goins, N.A. Petek, R.D. Mullins.
The team members studied these interactions in the specific case of nonmuscle Drosophila tropomyosin protein, Tm1A. They also compared some of these interactions in Tm1A to the same interactions in rabbit skeletal muscle tropomyosin, as other researchers previously have found that mammalian skeletal muscle tropomyosin is the least-effective Arp2/3 inhibitor.2
Data from dual-wavelength excitation produced by TIRF microscopy methodology when applied to single filaments is shown in Figure 3. This information shows that Tm1A preferentially binds near the pointed end of actin filaments. By comparing similar data that resulted from different experimental conditions, the researchers showed that pointed-end binding is dependent on the nucleotide state of the actin and the Tm1A concentration.
Although a complete evaluation of all of the research’s results, conclusions and wider implications falls outside the scope of this article, Mullins does summarize some of the key points. “Binding of cyto-skeletal tropomyosin to actin filaments turns out to be more complicated than previously appreciated. Both nucleation and spreading of tropomyosin are strongly influenced by the conformation of the actin filament and the presence of other regulatory proteins.” Mullins added that, based on TIRF-produced images and other collected data, “We have been able to propose a model where the cooperation of the severing activity of cofilin and tropomyosin binding helps establish the border between the lamellipod and lamellum.” The role of cofilin in the model referenced by Mullin is shown in Figure 4.
![These images summarize the role of cofilin in the model proposed by Hsaio et al. [ref].](https://www.photonics.com/images/Web/Articles/2015/9/8/Microscopy_Cofilin.png)
Figure 4. These images summarize the role of cofilin in the model proposed by Hsaio et al. [ref]. The branched actin network on the left shows the situation in the absence of cofilin, where tropomyosin binding is blocked by Arp2/3 branches. The branched actin network on the right illustrates that in the presence of cofilin, new pointed ends are created, which allows tropomyosin to bind. Once tropomyosin is bound, it protects the actin filaments from further cofilin severing, possibly resulting in the transition from the lamellipod to the lamellum. Courtesy of J.Y. Hsiao, L.M. Goins, N.A. Petek, R.D. Mullins.
In summation, TIRF microscopy is a well-established technique for imaging single molecular structures and protein complexes. This method also enables their respective dynamics to be observed in real time. By providing a method to rapidly switch between two or more excitation wavelengths, the latest lasers and laser-combining technologies are now enabling researchers to perform TIRF microscopy experiments with a greater number of separate labels. This capability is delivering unique insights into important and multifaceted processes in the study of cell biology.
Meet the author
Dan Callen is a product manager at Coherent Inc. in Santa Clara, Calif.; email: [email protected].
References
1. A.D. Edelstein et al. (2014). Advanced methods of microscope control using μManager software. J Biol Methods, Vol. 1, No. 2, e10.
2. J.Y. Hsiao et al. (2015). Arp2/3 complex and cofilin modulate binding of tropomyosin to branched actin networks. Curr Biol, pp. 1-10.
3. L. Blanchoin et al. (2001). Inhibition of the Arp2/3 complex-nucleated actin polymerization and branch formation by tropomyosin. Curr Biol, Vol. 11, No. 16, pp. 1300-1304.
4. J.H. Iwasa and R.D. Mullins (2007). Spatial and temporal relationships between actin-filament nucleation, capping and disassembly. Curr Biol, Vol. 17, No. 5, pp. 395-406.