Prashant Prebhat and Turan Erdogan, !%Semrock Inc.%!
The latest incarnation of the modern fluorescence microscope has led to a paradigm shift. This wave is about breaking the diffraction limit first proposed in 1873 by Ernst Abbe. The implications of this development are profound. So-called superresolution microscopy allows for the visualization of cellular samples with a resolution similar to that of an electron microscope, yet it retains the advantages of an optical fluorescence microscope. This means it is possible to uniquely visualize desired molecular species in a cellular environment – even in three dimensions and now in live cells – at a scale comparable to the spatial dimensions of the molecules under investigation.
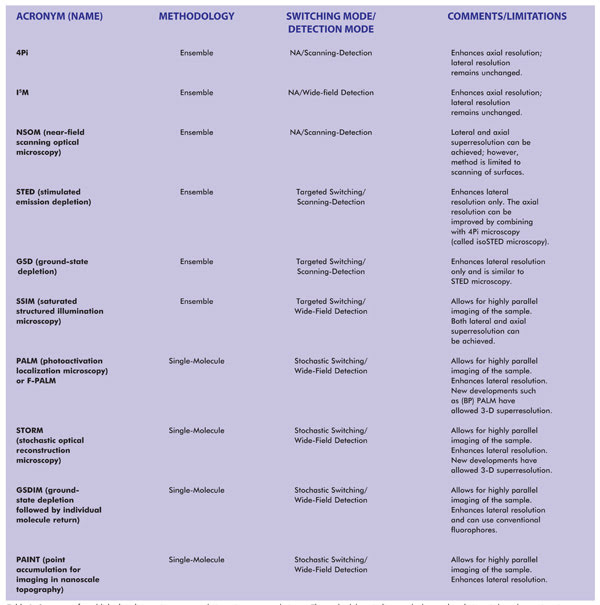
Table 1. Summary of established and emerging superresolution microscopy techniques. The methodology indicates whether each technique is based upon imaging of single molecules or imaging of an ensemble of molecules (which, in the limiting case, can also image a single molecule). NA = not applicable. Notes: SSIM is also referred to as SPEM (saturated pattern excitation microscopy). RESOLFT (reversible saturable/switchable optically linear fluorescence transition) is a generalized name for STED or SPEM. F-PALM (fluorescence PALM) is synonymous with PALM. Variants of PALM include TL-PALM (time-lapse PALM), PALMIRA (PALM with independently running acquisition), spt-PALM (single-particle-tracking PALM), iPALM (interferometric PALM) and biplane PALM, or (BP) PALM, a method that also enhances axial superresolution. Variants of STORM include: direct STORM (dSTORM), based on conventional fluorophores, and 3D-STORM.
So what is wrong with a conventional fluorescence microscope? To understand the answer, consider a green fluorescent protein (GFP) that is about 3 nm in diameter and about 4 nm long. When a single GFP molecule is imaged using a 100x objective, the size of the image should be 0.3 to 0.4 µm (100 times the size of the object). Yet the smallest spot that can be seen at the camera is about 25 µm, which corresponds to an object size of about 250 nm. Thus, there is a big disconnect between reality (the size of the actual fluorophore) and its perception (the image at the camera). The image suggests a much larger object size than is actually present.
Why is there a disconnect? The answer lies in the fundamental limitations imposed by the optics used to image such molecules. Conventional fluorescence microscopy uses a lens to focus a beam of light onto a spot. For example, a lens focuses the emission signal onto a CCD detector, or the illumination beam onto the sample, such as in a confocal scanning microscope. Performance of all such instruments is dictated by far-field optics (distances much greater than the wavelength of light).
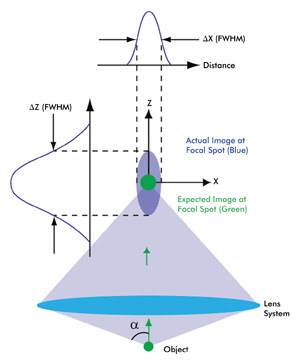
Figure 1. This schematic depicts the intensity distribution from a very small object (i.e., smaller than the wavelength of light). Because of diffraction, the actual image (spread over a 3-D region, violet) is much larger as well as elongated compared with the expected magnified image (green). The 3-D intensity distribution of the actual image is called the point spread function (PSF) of a microscope. A camera placed in the focal plane of the lens system – which includes the objective and the tube lens – captures a cross section of this 3-D intensity distribution. The cross-section profiles of the intensity distribution along the X and Z axes also are shown. As a result of circular symmetry of the PSF, ΔX = ΔY, a is one-half of the angular aperture of the objective lens. Various colors of the expected and actual images are used for the purpose of illustration. The schematic is not to scale.
In the far-field regime, diffraction plays a dominant role in image formation and, in fact, limits the smallest spot size that can be obtained at the focal point of a lens (Figure 1). Because of diffraction, a parallel beam of light is focused by a lens into a three-dimensional region near the focal point. This intensity distribution near the focal point is referred to as the point spread function (PSF) of a microscope and forms the basis of the resolution of a microscope. The full widths at half maximum (FWHM) of the PSF in the lateral (X-Y) plane and along the optical (Z) axis are given by
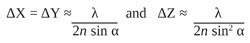
respectively, where λ is the wavelength of light, n is the refractive index of the medium in which light propagates, and α is one-half of the angular aperture of the objective lens.
Because of the nonzero value of the FWHM of the PSF, when two objects are located very close to each other, their images cannot be distinguished. The resolution of a microscope is the smallest distance between two objects that can be discerned by the microscope. Abbe proposed that the lateral resolution (X-Y plane) of an optical microscope is given by
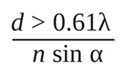
which is based upon the observation of a PSF (see Figure 2A).
An implication of limited resolution is that fine structures cannot be discerned. For example, consider microtubules that serve as structural scaffolds within cells and are about 25 nm in diameter. A 250-nm-diameter (diffraction-limited) image of a microtubule revealed by conventional microscopy actually may represent a bundle of several microtubules that cannot be distinguished from one another. In this case, enhanced resolution would provide additional information about the cellular architecture. Superresolution also enables the study of membrane heterogeneity and the dynamics of protein assembly – studies that benefit from observing single molecules.
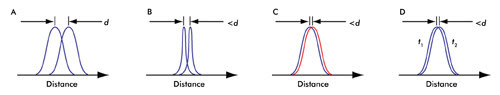
Figure 2. (A) In a conventional microscope, the smallest distance that can be discerned is given by d (see text). (B) Changing the PSF of the microscope enhances the resolution. (C) Abbe’s resolution criterion is not limited by color. (D) Time distinction (see text) is a basis of all the recent superresolution techniques; t1 and t2 are different instances of time.
Beating the diffraction limit
Superresolution microscopy is about enhancing the diffraction-limited resolution of a microscope. Near-field scanning optical microscopy is a superresolution microscopy technique based on near-field optics. Using this technique, a spot size of only a few nanometers can be resolved. However, this technique is limited to the study of surfaces only.
Initial attempts at enhancing the resolution of a microscope, using far-field optics, involved designing objective lenses of higher numerical aperture (NA) where NA = n sin α. Examples of superresolution microscopy techniques that use this approach include “4Pi” and “I5M” microscopy (Figure 2B).
Changing the PSF (as in “4Pi” and “I5M”) is not the only way to enhance the resolution. Because Abbe’s resolution criterion applies to a given color, the resolution of a fluorescence microscope should not be limited when two closely spaced point sources emit light in different colors. Therefore, in theory, if all the molecules of a sample could be labeled with a different color, then a resolution better than diffraction-limited imaging could be achieved1 (Figure 2C). Förster resonance energy transfer microscopy takes advantage of this principle.
Abbe’s resolution criterion also does not impose a limit on the resolution when two closely spaced point sources are imaged at different times; i.e., imagine two PSFs that cannot be otherwise distinguished but where each is observed at a different time (Figure 2D). This methodology of “sequential” imaging forms the basis of the most recent and successful superresolution microscopy techniques.
Recent superresolution microscopy techniques
The mechanism by which time distinction is achieved can be explained by the electronic transition states of a fluorophore (Figure 3A). In conventional fluorescence microscopy, a fluorophore absorbs energy from the excitation light and almost instantaneously releases the emission signal. To achieve time distinction – which ultimately provides spatial discrimination – nonlinear relationships between excitation and emission of a fluorophore are exploited. By virtue of these nonlinear relationships, specific fluorophore molecules can be switched on or off.
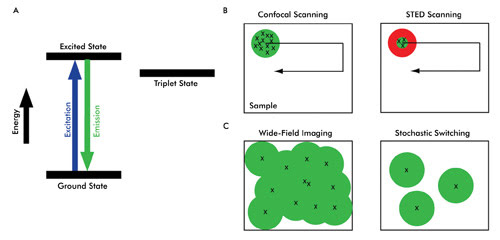

Depending upon how time distinction is achieved, superresolution microscopy techniques can be broadly categorized into two main approaches.1 In one, called “targeted switching and readout,” the illumination volume in a fluorescent sample is confined to a small region, which is much smaller than the diffraction-limited spot size. Stimulated emission depletion (STED) microscopy is based on this approach (see Figure 3B). Knowledge about the illumination spot size is used to generate a superresolution image. The other approach, “stochastic switching and readout,” uses stochastic variation associated with switching fluorophore molecules on or off (Figure 3C).
The first superresolution microscopy technique that used this nonlinear relationship is STED. In conventional point-scanning confocal systems, all the fluorophore molecules within a diffraction-limited excitation volume are excited and emit the fluorescence signal simultaneously (Figure 3B, left). Moving the scanning beam over the sample creates a composite image.
The basic implementation of stimulated emission depletion microscopy is similar in terms of point-by-point scanning to generate a single image. However, in STED microscopy, the emission signal is generated from a much smaller volume than from that of a conventional confocal scanner. The laser-scanning illumination (excitation pulse) spot is overlapped by another beam called the STED beam, which has an annular, or “doughnut,” shape and is of a longer wavelength than that of the scanning beam. Because of stimulated emission, the effective emission spot size is reduced to well below the diffraction limit (Figure 3B, right), which improves the lateral resolution of the microscope.
Saturated structured illumination microscopy is another superresolution technique based on targeted switching. The superresolution effect in this method is achieved by illuminating the fluorophores with patterned excitation light of saturating intensity. Sophisticated mathematical analysis of the data acquired in wide-field detection mode is used to generate a superresolution image.
Consider another radically different approach for achieving time distinction: Rather than controlling the illumination spot, what if it were possible to switch single molecules of fluorophores on (i.e., to an excitable state that can be imaged) and off (to a state that cannot be imaged)? At a given time, only a small population of fluorophores is turned on for imaging (Figure 3C, right). This subset is then turned off after imaging.
Repeating the process of switching on a small population of fluorophore molecules, imaging and then turning them off generates a sequence of images. Finally, all these images with artificially smaller spot sizes are superimposed to arrive at a superresolution image.
Photoactivation localization microscopy (PALM, also referred to as F-PALM for fluorescence PALM) and stochastic optical reconstruction microscopy (STORM) were the first techniques to exploit this principle of stochastic switching of fluorophores to generate superresolution images. Specially designed fluorophores that are photoactivatable or photoswitchable were first used in the development of PALM and STORM.
GSDIM (ground-state depletion followed by individual molecule return) is another example of stochastic switching and readout. In this technique, ordinary fluorophores are initially turned off – e.g., by driving them into the long-lived triplet states (Figure 3A) – and then, as individual molecules return to the ground state stochastically, a readout scheme similar to photoactivation localization microscopy and stochastic optical reconstruction microscopy enables superresolution microscopy.
Because of the numerous possibilities of imaging configurations, the requirements for optical filters in superresolution imaging often are best met by custom-selecting filters for a given system. A combination of filters that can be used for photoactivation and imaging of Dronpa is shown in Figure 4. A special characteristic of the dichroic beamsplitter shown in this figure is its wide reflection band, which is compatible with both the activation (~405-nm) and the readout (~488-nm) lasers. High transmission of the emission filter ensures maximum signal collection from a limited population of fluorophores imaged at a given time, enhancing overall throughput.
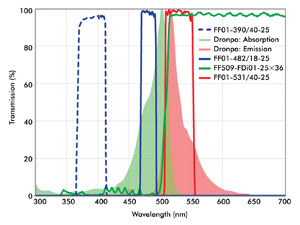
Figure 4. Shown are optical filters useful for imaging of Dronpa. The wide reflection band of the dichroic ensures that both the activation light (e.g., a 405-nm laser) and imaging beam (488-nm laser) are efficiently reflected and rejected in transmission. A compatible emission filter provides high blocking of both the activation and imaging lasers.
An essential step in single-molecule-based superresolution imaging techniques (stochastic switching and readout) is to accurately localize individual fluorophore molecules. With higher accuracy of localization, higher superresolution is achieved. Because the accuracy of localization of a given fluorophore increases dramatically with the number of photons acquired from a given fluorophore molecule, highly efficient optical filters play an increasingly important role in superresolution microscopy.
Perspective
Excitement about superresolution imaging techniques is evident from the surprisingly rapid commercialization of turnkey instruments. For example, Leica was first to launch a commercially available superresolution microscope using stimulated emission depletion and a GSDIM system at the time of writing this article. Zeiss and Nikon now also offer superresolution microscopes that use PALM and STORM, respectively, as well as variants of structured illumination microscopy.
The commercial availability of superresolution instruments will enable widespread research at unprecedented resolution. Research still continues to enhance the performance of superresolution techniques in terms of overall throughput so that they can be used even for the study of fast cellular dynamics in live cells.
And efforts are under way to enhance the superresolution not only laterally but also axially. Rather than having to develop and use specialized fluorophores, many researchers have demonstrated the advantages of using standard fluorophores, expanding the applicability of super-resolution techniques.
Reference
1. Special Feature: Method of the Year (January 2009). Super-resolution microscopy: breaking the limits. Nature Methods, Vol. 6, No. 1.
Meet the authors
Dr. Prashant Prabhat is an applications scientist and Dr. Turan Erdogan is co-founder and CTO, both at Semrock Inc., a unit of Idex Corp.; e-mail: [email protected]; [email protected].