Dr. Stuart Yin, Pennsylvania State University
High-speed, multichannel fluorescence confocal imaging can be achieved
by encoding the spatial location information into the frequency domain.
Fluorescence confocal microscopy is an important tool for studying
live biological cells, and it can reject out-of-focus fluorescence so that an image
with a high signal-to-noise ratio can be obtained over the scattering biomedical
media.
To understand cellular systems, it is necessary
to observe fast biological processes — such as cardiac myocyte contraction
— in real time. Multichannel methods can increase imaging speed; however,
although several have been developed, they are difficult to use directly with fluorescence
confocal microscopy.
For example, wavelength division multiplexing
cannot be employed because the wavelength of the fluorescence emission is determined
by the fluorescent label, which is independent of the wavelength of the incident
excitation beam.1,2
Another major challenge comes from
the weak fluorescence emission signal. To effectively detect a weak signal at fast
speeds, a highly sensitive photomultiplier tube usually is employed. In general,
a photomultiplier tube is a single-pixel detector. However, many multiplexing techniques
— such as those using microlens and pinhole arrays — require a highly
sensitive imaging detector (that is, an array of detectors).3
Although photomultiplier tube arrays
recently have been used as the detection modules in commercial systems, they usually
have a limited number of pixels (e.g., 32), and, furthermore, these modules are
very expensive.
On the other hand, the sensitivity
of the CCD-based detector is limited by the imaging speed. Commonly available CCD
imaging detectors used for multichannel confocal microscopy operate at about 30
fps, which is not fast enough to monitor many of the dynamics that occur in cells.
Our group at Pennsylvania State University
is studying a long-standing problem in cardiac research: How to obtain the transient
3-D distribution of calcium ions in a cardiac myocyte during excitation-contraction.
This type of study requires high spatial (~100 nm) and temporal (~1
ms) resolutions.
Frequency division multiplexing
To overcome the limitations of existing fluorescence
confocal microscopy, we recently developed a frequency division multiplexed fluorescence
confocal microscope.4 The device provides high spatial resolution as well as temporal
resolution up to the nanosecond range, limited only by the lifetime of the fluorophores
and the response time of the photomultiplier tube.
This technology can be applied to detection
modules based on either single or arrayed photomultiplier tubes, significantly increasing
the number of multiplexing channels for both. This enables high-resolution fluorescence
imaging in real time.
To illustrate the working principle
of a frequency division multiplexed fluorescence confocal microscope, consider a
two-channel version of the device in which a 488-nm incident beam — emitted
from an argon-ion laser from Coherent Inc. of Santa Clara, Calif. — is divided
into two by a beamsplitter (Figure 1).
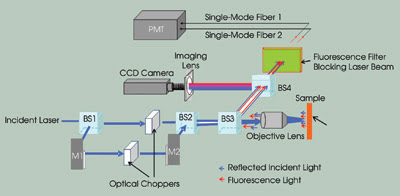
Figure 1. This illustration shows a two-channel frequency division
multiplexed fluorescence confocal microscope. (BS: beamsplitter; M: mirror; PMT:
photomultiplier tube). Images reprinted with permission of Biophysical Journal.
The intensities of the two beams are
individually modulated at different carrier frequencies —
ω;1
= 2 π f1 and ω2 = 2 π f2, respectively — and then recombined
by means of another beamsplitter. After passing through a third beamsplitter, the
two modulated beams are further coupled into the back aperture of a 60x, 1.4-NA
objective lens from Nikon Corp. at slightly different angles. The modulated Gaussian
beams are focused at two locations on the sample by the objective, forming two spots.
The distance between the two spots
is adjustable by tuning the relative angle between two incident beams, which can
be realized by adjusting the reflection angles of the mirrors. The fluorescence
emission from the sample at the two focusing spots is collected by the same objective
lens and reflected to the detection system by a beamsplitter.
To view the sample and the location
of the focusing spots, a fourth beamsplitter is added in the detection portion
of the system. The reflected light from this beamsplitter enters a CCD imaging system,
and the light that it transmits passes though a bandpass filter that blocks the
excitation laser beams and transmits only the fluorescence light.
The transmitted fluorescence light
beams from the two focusing spots are then focused into two single-mode optical
fibers by two objective lenses, with each fiber corresponding to one spot. The single-mode
fibers are used as pinholes to filter out the out-of-focus light and to achieve
confocal imaging5,6 and, conveniently, to couple the light beams into a single photomultiplier
tube.
The intensity detected by the photomultiplier
tube is the summation of these two modulated intensities. The output from the photomultiplier
tube is connected to a data acquisition board, which converts the analog electric
signal into a digital signal. The data is sent to a microcomputer that processes
it by taking the Fourier transform of the detected signal. In the frequency domain,
there is no overlap between the two signals. Thus, one can easily classify
both signals in the frequency domain.
The worst case scenario for crosstalk
noise among various frequency channels (that is, the highest amount of crosstalk)
happens when the two laser spots are focused onto the same location. In this case,
the signal separation can be realized only from the difference carrier frequency.
The ideal situation (no crosstalk noise)
occurs when the lowest carrier frequency and the separation between adjacent frequencies
are twice the signal frequency (i.e., Nyquist sampling theorem). However, because
other noise (e.g., from the detector) exists, there will be crosstalk among laser
channels.
Without losing the generality, if one
assumes that the noise is Gaussian, then the bandwidth of the noise spectrum can
be estimated by observing the detected signal over a finite time interval.7 Then,
one can select the frequency and the frequency difference between adjacent channels
at least twice this estimated spectral bandwidth, which will ensure low cross-talk
noise.
Limit on number of channels
The maximum number of the frequency division multiplexed
channels is limited by the response time of the fluorescence emission and the photomultiplier
tube detector and by the dynamic range of the photodetector. The temporal resolution
of this method also is determined by the response time of the fluorescence emission
and the photodetector, which is on the order of 10 ns. Because this resolution is
usually adequate to analyze the dynamic behavior of living cells, the response-time-limited
number of channels can be estimated by dividing the signal’s temporal bandwidth
(1 ms) with temporal resolution (2 x 10 ns = 20 ns), which is as high as 5 x 104.
On the other hand, the dynamic-range-limited
number of channels may be estimated by assuming that the useful dynamic range of
the photodetector is 30 dB (that is, 1000 in the linear scale, a realistic number)
and that the required dynamic range from each frequency channel is 10 dB (10 in
the linear scale).
In this case, the dynamic-range-limited
number of channels is 100 (1000/10). Thus, the maximum number of frequency division
multiplexed channels is mainly determined by the dynamic range of the photodetector.
The total number of channels can be
increased further by employing a photomultipler tube array. A 32-channel array is
commercially available, and combining our frequency division multiplexing technique
with it can make the total number of channels as large as 100 x 32 = 3200. This
is good enough for many real-time confocal imaging applications.
The spatial resolution of this frequency
division multiplexed fluorescence confocal microscope is the same as conventional
fluorescence confocal microscopes.
To verify the feasibility of our technique,
we set up a two-channel experimental demonstration system (Figure 2). A 488-nm argon-ion
laser was used as the excitation source. The channels were modulated at 350 and
400 Hz, respectively, by two optical choppers that are conventionally used for lock-in
amplifiers. A 60x, 1.4-NA objective from Nikon was used as the focusing lens, which
focused the two modulated beams onto the sample.

Figure 2. Researchers used the two-channel frequency division multiplexed fluorescence confocal microscope
shown here to study intracellular calcium-ion concentrations.
A living rat cardiac myocyte that had
an average dimension of ~100 μm long x 20 μm in diameter was used
as the sample. The fluorescent calcium-ion indicator fluo-4 AM ester was loaded
in freshly isolated adult rat myocytes (1.8 μM, 30 min at 37 °C), which
then emitted green light (520 to 540 nm) when they were illuminated by the 488-nm
blue light (Figure 3). The fluorescence from the two focusing spots was collected
by the same objective lens. A bandpass filter (520 to 540 nm) blocked the excitation
laser beam. The transmitted fluorescence emissions were coupled into two single-mode
fibers by two objectives. A Hamamatsu photomultiplier tube with a response time
of ~10 ns was used to detect the fluorescence emissions.
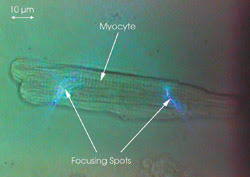
Figure 3. A living rat cardiac myocyte has two spots focused on it.
Real-world test
As an example of real-world applications of this
high-speed, multichannel fluorescent confocal microscopy technology, the experimental
system was used to study the simultaneous changes in calcium-ion concentration in
a living cardiac myocyte. It is well known that calcium ions occupy a central role
in cardiac excitation-contraction coupling.8-10
Recent high-resolution imaging suggests
that, even though only a small number of calcium ions enter the cell, the local
calcium concentration in this “cleft” between the calcium channel and
the ryanodine receptor is likely to be substantially higher than that measured in
the myoplasm. Other indirect evidence also suggests the existence of a “submembranous”
domain in which the concentrations of calcium and sodium ions are significantly
different from those in bulk myoplasm — which has major ramifications for
understanding the mechanisms of excitation-contraction coupling in the cardiac myocyte.
Despite the importance of answering
the question of whether there is a change in local concentrations of calcium and
sodium ions in the submembranous domain during the excitation-contraction coupling,
very few studies have provided direct measurements because of the limited temporal
resolution of conventional fluorescence confocal microscopes.
Using frequency division multiplexed
fluorescence confocal imaging technology, we have tracked simultaneous changes in
calcium concentrations in the submembranous domain and in the bulk cytosol in a
living cardiac myocyte during an action potential.
Freshly isolated adult rat myocytes
were loaded with fluo-4. The confocal laser excitation beams were directed separately
onto the cell membrane region as well as into the bulk cytosol. To ensure that
one of the modulated laser excitation beams was localized at or near the cell membrane,
while the other was in the bulk cytosol, we “doubly” labeled the myocyte
with a fluorescent membrane potential indicator (di-4-ANEPPS), which distributes
to the charged plasma membrane (surface membranes and transverse tubules) with little
to no signal in the cytosol.
Because of the overlap between the
emission spectrum of di-4-ANEPPS and the fluo-4, a bandpass filter (520 to 540 nm)
and a long-pass filter (550 nm) were applied separately to discriminate between
the two emitted fluorescence signals. Because fluo-4 is excited by a single wavelength,
its fluorescence intensity is proportional to that of the excitation light, the
optical light path, the fluorescent probe concentration as well as the free-calcium
concentration.
To ensure that the intensity of fluo-4
reflected the free-calcium concentration in the region interrogated, the intensities
of the excitation beams were at two focusing spots and were further balanced by
adjusting one of the beams until both had equal intensity. Second, all of the optical
light paths were fixed. Finally, the uniformity of the concentration of fluo-4 was
realized by following the standard procedure (that is, loading the fluo-4 at 37
°C at least 15 min before the start of the experiment).
To minimize the motion artifact of
the myocyte when applying the stimulating electric field, we used cytochalasin D
to immobilize the cell while preserving transients in cytosolic calcium concentrations.
We stimulated the myocyte to contract using field electrodes at 1 Hz, continuously
sampling the fluo-4 signals for 5 to 10 s.
The detected data was processed according
to the described procedure. Figure 4 shows the measured temporal variation of both
the cytosolic calcium concentration around the membrane region and the calcium concentration
in the bulk cytosol of the same myocyte.
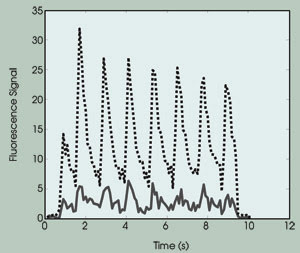
Figure 4. The intracellular calcium-ion concentration exhibits beating curves. The solid curve
shows the fluorescence emission from the bulk region, and the dotted curve shows
the fluorescence emission from the membrane region.
From this experimental result, we draw
the following conclusions: First, the calcium-ion concentration changes during the
cell contraction. Second, the rate of change is the same as the excitation rate
(i.e., 1 Hz; the beating behavior). Third, the calcium concentration at the submembranous
region was approximately five to six times higher than that measured at the bulk
cytosol region, which was consistent with predictions.8,9,10
Meet the author
Stuart (Shizhuo) Yin is a professor in the electrical
engineering department at Pennsylvania State University in University Park; e-mail:
[email protected].
References
1. J. Tearney et al (1998). Spectrally encoded
confocal microscopy. OPT LETT, pp. 8214-8221.
2. Z. Yaqoob and N. Riza (2002). Free-space
wavelength-multiplexed optical scanner demonstration. APPL OPT, pp. 5568-5573.
3. K. Fujita et al (2000). Confocal
multipoint multiphoton excitation microscope with microlens and pinhole arrays.
OPT COMM, pp. 7-12.
4. F. Wu et al (2006). Frequency division
multiplexed multi-channel high speed fluorescence confocal microscope, BIOPHYS
J, pp. 2290-2296.
5. M. Gu et al (1991). Image formation
in a fiber-optical confocal scanning microscope, J Opt Soc Am A, Vol. 8.
6. S. Kimura (1991). Confocal scanning
optical microscope using single-mode fiber for signal detection, APPL OPT,
pp. 2143-2150.
7. J. Proakis et al (1992). Advanced
Digital Signal Processing, Chapter 8: Power Spectrum Estimation, Macmillan Publishing
Co., New York, p. 473.
8. D. Bers (2002). Cardiac excitation-contraction
coupling, NATURE pp. 198-205.
9. J. Cheung et al (2004). Exercise
training improves cardiac function post-infarction: Special emphasis on recent controversies
on Na+/Ca2+ exchanger. EXERC SPORT SCI REV, pp. 83-89.
10. D. Scriven et al (2000). Distribution
of proteins implicated in excitation-contraction coupling in rat ventricular myocytes.
BIOPHYS J, pp. 2682-2691.