EMCCD technology aids sequencing technique.
Dr. George M. Church, Dr. Gregory J. Porreca, Richard C. Terry, Harvard Medical School, and Maridel Lares, Hamamatsu Corp.
The key to personalized medicine is understanding the role that an individual’s genes play in his or her health. To reap the benefits of personalized medicine, the billions of nucleotide bases that comprise an individual’s genome must be sequenced — and done so at an affordable price. This means lowering the price tag from the $3 billion spent on the Human Genome Project to $100,000 and, eventually, to $1000. Reaching this goal will require a fundamentally different approach to DNA sequencing that delivers significantly more bases of accurate sequence per dollar than do current sequencers.
The $1000 goal is still far in the future, but the efforts of many researchers and companies have brought us closer to the $100,000 mark. Among the new sequencing systems developed is the Polonator from George M. Church’s laboratory at Harvard Medical School in Boston, which uses a cyclic-array sequencing technique.1
‘Polony‘ sequencing
Today’s sequencing method -- based on concepts from Frederick Sanger and Walter Gilbert’s work in the 1970s2,3 – uses fluorescent-labeled nucleotide bases (adenine, cytosine, guanine and thymine) and capillary electrophoresis to determine the order of bases in a DNA strand. Since 1988, researchers have multiplexed DNA sequencing as a way to lower costs.4 This led to the development of polymerase colonies (or “polonies”) on glass slides in 19995,6 and to polonies on beads in 2005.7,8,9
Creating polonies is a process of copying fragments of DNA strands in vitro, unlike conventional methods that use bacterial colonies. The technique is used in the Polonator D.005, a sequencing system developed in Church’s laboratory that comprises a Nikon microscope with automated X-Y stage and Z-focus, xenon illumination, an autosampler for chemical reagents, a Hamamatsu electron-multiplying (EM) CCD camera and a computer.
Polony DNA sequencing7,10 arranges via ligation of fluorescent probes on microbeads bearing clonally amplified DNA templates. These are attached to a microscope cover glass in a flow cell, which is mounted on an automated fluorescence microscope. Successive cycles of fluorescence probing to interrogate a position within the unknown DNA template enable the DNA sequence of each bead to be compiled. Each cycle of ligation is followed by four-color epifluorescence imaging to determine the wavelength of fluorescence emission from each bead. In each cycle, a given bead will fluoresce in only one of the possible four channels, indicating whether that bead has an adenine, a cytosine, a guanine or a thymine at the position being interrogated (Figure 1).

Figure 1. This false-color composite shows fluorescence image data from one position on the Polony sequencing bead array (about 1/20,000 of one cycle of data). Four images are acquired and superimposed; the identity of the base being sequenced on each bead can be determined by its color — e.g., red: adenine, green: cytosine, blue: guanine, yellow: thymine. Approximately 400 GB of images are collected per flow cell per run (of 26 cycles) and converted into DNA sequence by our software.
EMCCD camera
Detecting the weak fluorescence of the beads requires a highly sensitive camera, while the high throughput needed for faster sequencing requires high speed (30 frames per second). To address both issues, a highly sensitive and high-speed EMCCD camera from Hamamatsu was chosen. An EMCCD is a type of CCD with a special multiplication register between the horizontal register and the output node. Each stage in the multiplication register produces a gain in the signal through impact ionization (Figure 2).
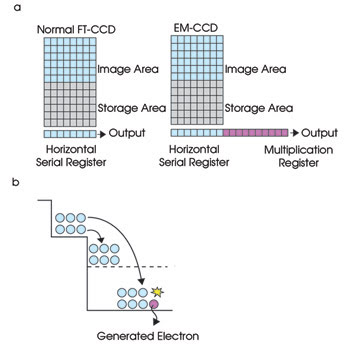
Figure 2. (a) In an EMCCD, a special multiplication register generates a gain in the signal through a process called impact ionization. (b) Impact ionization creates extra electron-hole pairs, multiplying the electrical signal in an EMCCD. FT = frame transfer.
For impact ionization to occur, a higher-than-normal voltage (about 30 to 40 V) is applied to the multiplication register. During charge transfers in this register, the electrons are given enough energy by the increased electric field strength to free electrons bound to silicon atoms and to add to the charge. Essentially, a packet of charge picks up extra passengers (secondary electrons) while being transferred from one pixel to the next, amounting to an increase in electrical signal. The probability of impact ionization is very small (about 1.0 to 1.6 percent) at each stage, but having 400 to 600 stages produces a significant increase in signal, referred to as electron-multiplication gain, ranging from four to 2000 times.
The gain is dependent on temperature and external voltage, so stability is critical. Any fluctuation in the temperature or applied voltage causes alterations in the gain factor, affecting repeatability from image to image. A highly stable cooling system and the stable application of voltage through excellent electronics design therefore play a vital role in an EMCCD camera’s performance.
The sources of noise in an EMCCD are dark current, spurious charge from clocking, and readout and excess noise. The dark current and spurious charge noise are minimized by deep cooling and proper design of electronics, respectively. Excess noise is that associated with the multiplication of the signal in the multiplication register.
High-speed imaging
Signal-to-noise ratio is important in high-speed imaging because two conditions occur that affect both. The short integration time reduces the amount of light collected, resulting in a weaker electrical signal, while the faster readout speed introduces more readout noise.
These two conditions could prove problematic in CCD cameras without an internal gain; the cameras would not detect extremely weak signals during fast readout speeds. But the lower signal and higher readout noise are less troublesome for an EMCCD camera because of its high gain. The camera can multiply weak signals to be detectable above the readout noise.
A completely re-engineered commercial-grade version of the Polonator (version G.007) is now available from Danaher Motion-Dover of Westborough, Mass. The only components retained from the D.005 version are the light source and the Hamamatsu EMCCD camera.
Analogous to the IBM PC introduced in 1981, the Polonator is intentionally an open-architecture system (hardware, software and wetware) to encourage a diverse commercial and academic ecosystem and to drive down costs to make genomics affordable to everyone. Just as the personal computer led to applications well beyond its utility in 1981, current DNA-sequencing technology should lead us into the exciting age of personalized genomes, individualized medicine and beyond.
Meet the authors
George M. Church is a professor of genetics at Harvard Medical School in Boston; http://arep.med.harvard.edu/gmc/email.html.
Gregory J. Porreca is a member of Church’s laboratory at Harvard Medical School; e-mail: [email protected].
Richard C. Terry is a member of Church’s laboratory at Harvard Medical School; e-mail: [email protected].
Maridel Lares is a technical writer at Hamamatsu Corp. in Bridgewater, N.J.; e-mail: [email protected].
References
1. J. Shendure et al (May 2004). Advanced sequencing technologies: methods and goals. NAT REV GENET, pp. 335-344.
2. F. Sanger et al (Dec. 1, 1977). DNA sequencing with chain-terminating inhibitors. PNAS, pp. 5463-5467.
3. A.M. Maxam and W. Gilbert (Feb. 1, 1977). A new method for sequencing DNA. PNAS, pp. 560-564.
4. G.M. Church and S. Kieffer-Higgins (April 8, 1988). Multiplex DNA sequencing. SCIENCE, pp. 185-188.
5. R.D. Mitra and G.M. Church (1999). In situ localized amplification and contact replication of many individual DNA molecules. NUCLEIC ACIDS RES, Vol. 27, Issue 24, e34.
6. R.D. Mitra et al (Sept. 1, 2003). Fluorescent in situ sequencing on polymerase colonies. ANAL BIOCHEM, pp. 55-65.
7. J. Shendure et al (Sept. 9, 2005). Accurate multiplex polony sequencing of an evolved bacterial genome. SCIENCE, pp. 1728-1732.
8. M. Margulies et al (Sept. 15, 2005). Genome sequencing in microfabricated high-density picolitre reactors. NATURE, pp. 376-380.
9. D. Dressman et al (July 22, 2003). Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variation. PNAS, pp. 8817-8822.
10. J.B. Kim et al (June 8, 2007). Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. SCIENCE, pp. 1481-1484.